





Oprócz technologii, synteza glikozydów zawsze była przedmiotem zainteresowania nauki, ponieważ jest to bardzo powszechna reakcja w naturze. Najnowsze prace Schmidta, Toshimy i Tatsuty, a także liczne cytowane w nich pozycje, omawiają szeroki zakres potencjalnych możliwości syntezy.
W syntezie glikozydów składniki wielocukrowe są łączone z nukleofilami, takimi jak alkohole, węglowodany lub białka. Jeśli wymagana jest selektywna reakcja z jedną z grup hydroksylowych węglowodanu, wszystkie pozostałe funkcje muszą zostać zabezpieczone w pierwszym etapie. Zasadniczo procesy enzymatyczne lub mikrobiologiczne, ze względu na swoją selektywność, mogą zastąpić złożone etapy chemicznej ochrony i deprotekcji, aby selektywnie chronić glikozydy w określonych regionach. Jednak ze względu na długą historię alkiloglikozydów, zastosowanie enzymów w syntezie glikozydów nie zostało szeroko zbadane i zastosowane.
Ze względu na wydajność odpowiednich układów enzymatycznych i wysokie koszty produkcji, synteza enzymatyczna alkilopoliglikozydów nie jest jeszcze gotowa do modernizacji do poziomu przemysłowego; preferowane są metody chemiczne.
W 1870 roku MAcolley opublikował raport na temat syntezy „acetochlorhydrozy” (1, rysunek 2) poprzez reakcję dekstrozy (glukozy) z chlorkiem acetylu, co ostatecznie dało początek historii szlaków syntezy glikozydów.
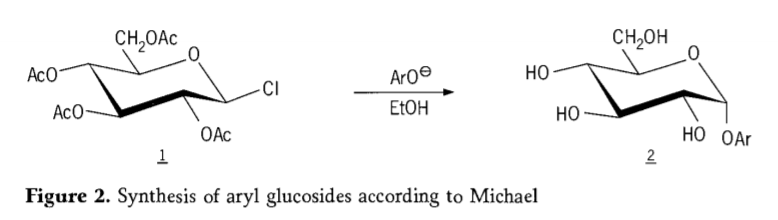
Później odkryto, że halogenki tetra-0-acetylo-glukopiranozylu (acetohaloglukozy) są użytecznymi związkami pośrednimi w stereoselektywnej syntezie czystych alkiloglukozydów. W 1879 roku Arthur Michael zdołał wytworzyć określone, krystalizujące glikozydy arylowe z związków pośrednich Colleya i fenolanów (Aro-, rysunek 2).
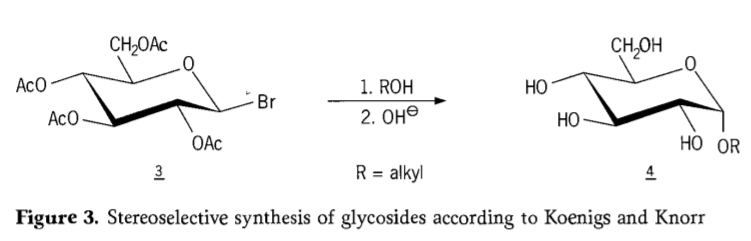
W 1901 roku Michael syntezował szeroką gamę węglowodanów i aglikonów hydroksylowych, kiedy W. Koenigs i E. Knorr wprowadzili ulepszony stereoselektywny proces glikozydacji (rysunek 3). Reakcja obejmuje podstawienie SN2 przy węglu anomerowym i przebiega stereoselektywnie z inwersją konfiguracji, wytwarzając na przykład α-glukozyd 4 z β-anomeru pośredniej aceobromoglukozy 3. Synteza Koenigsa-Knorra odbywa się w obecności promotorów srebra lub rtęci.
W 1893 roku Emil Fischer zaproponował fundamentalnie odmienne podejście do syntezy alkiloglukozydów. Proces ten jest obecnie znany jako „glikozydacja Fischera” i obejmuje katalizowaną kwasem reakcję glikoz z alkoholami. Każda relacja historyczna powinna jednak uwzględniać również pierwszą próbę A. Gautiera z 1874 roku, polegającą na przekształceniu dekstrozy w bezwodny etanol w obecności kwasu solnego. Z powodu mylącej analizy pierwiastkowej Gautier uważał, że otrzymał „diglukozę”. Fischer później wykazał, że „diglukoza” Gautiera to w rzeczywistości głównie etyloglukozyd (rysunek 4).
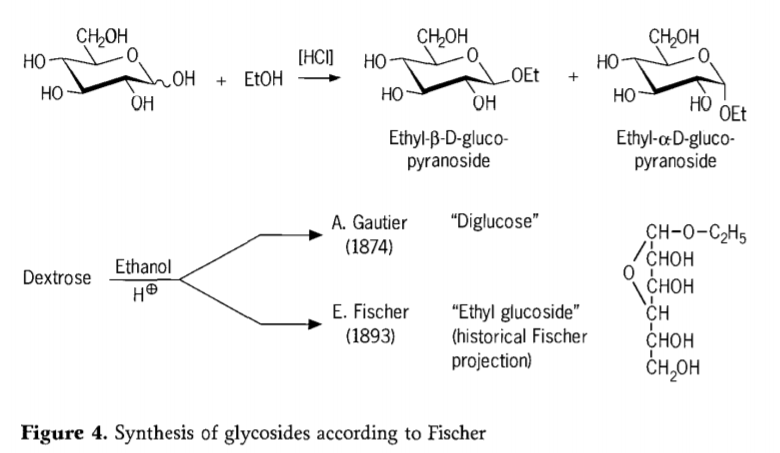
Fischer poprawnie zdefiniował strukturę etyloglukozydu, co widać na podstawie zaproponowanego historycznego wzoru furanozydowego. W rzeczywistości produkty glikozydacji Fischera są złożonymi, głównie równowagowymi mieszaninami α/β-anomerów i izomerów piranozydu/furanozydu, które zawierają również losowo połączone oligomery glikozydowe.
W związku z tym, izolowanie pojedynczych cząsteczek z mieszanin reakcyjnych Fischera nie jest łatwe, co w przeszłości stanowiło poważny problem. Po pewnym udoskonaleniu tej metody syntezy, Fischer zastosował następnie syntezę Koenigsa-Knorra w swoich badaniach. Wykorzystując tę metodę, E. Fischer i B. Helferich jako pierwsi opisali w 1911 roku syntezę długołańcuchowego alkiloglukozydu wykazującego właściwości surfaktantu.
Już w 1893 roku Fischer trafnie zauważył istotne właściwości alkiloglikozydów, takie jak ich wysoka odporność na utlenianie i hydrolizę, zwłaszcza w silnie alkalicznych środowiskach. Obie te cechy są cenne dla alkilopoliglikozydów w zastosowaniach jako środki powierzchniowo czynne.
Badania nad reakcją glikozydacji wciąż trwają, a w niedawnej przeszłości opracowano kilka interesujących metod syntezy glikozydów. Niektóre procedury syntezy glikozydów przedstawiono na rysunku 5.
Ogólnie rzecz biorąc, procesy glikozydacji chemicznej można podzielić na procesy prowadzące do złożonych równowag oligomerowych w wymianie glikozydowej katalizowanej kwasem.
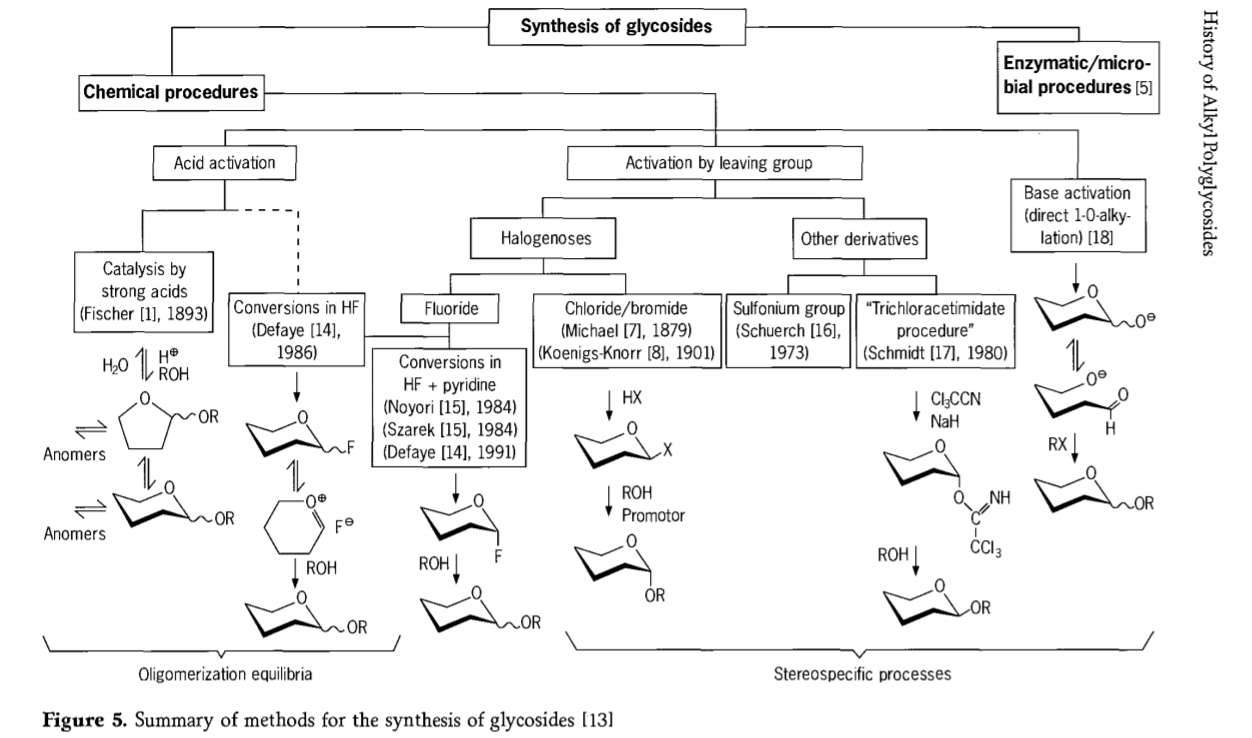
Reakcje na odpowiednio aktywowanych substratach węglowodanowych (reakcje glikozydowe Fischera i reakcje fluorowodoru (HF) z niezabezpieczonymi cząsteczkami węglowodanów) oraz kontrolowane kinetycznie, nieodwracalne i głównie stereotaktyczne reakcje podstawienia. Drugi rodzaj procedury może prowadzić do powstawania pojedynczych cząsteczek, a nie złożonych mieszanin reakcji, zwłaszcza w połączeniu z technikami grup konserwatywnych. Węglowodany mogą pozostawiać grupy na ektopowym atomie węgla, takie jak atomy halogenu, grupy sulfonylowe lub grupy trichloroacetimidowe, lub być aktywowane przez zasady przed przekształceniem w estry trifluorometanosulfonianowe.
W szczególnym przypadku glikozydacji we fluorowodorze lub w mieszaninach fluorowodoru i pirydyny (poli[fluorowodorek pirydyniowy]), fluorki glikozylu powstają in situ i są płynnie przekształcane w glikozydy, na przykład z alkoholami. Wykazano, że fluorowodór jest silnie aktywującym, niedegradującym środowiskiem reakcji; obserwuje się autokondensację (oligomeryzację) w stanie równowagi, podobną do procesu Fischera, chociaż mechanizm reakcji jest prawdopodobnie inny.
Chemicznie czyste alkiloglikozydy nadają się jedynie do bardzo specyficznych zastosowań. Na przykład, alkiloglikozydy były z powodzeniem stosowane w badaniach biochemicznych do krystalizacji białek błonowych, takich jak trójwymiarowa krystalizacja poryny i bakteriorodopsyny w obecności oktylo-β-D-glukopiranozydu (dalsze eksperymenty oparte na tej pracy doprowadziły do przyznania Deisenhoferowi, Huberowi i Michelowi Nagrody Nobla w dziedzinie chemii w 1988 roku).
W toku rozwoju alkilopoliglikozydów, metody stereoselektywne były stosowane w skali laboratoryjnej do syntezy różnorodnych substancji modelowych i badania ich właściwości fizykochemicznych. Ze względu na ich złożoność, niestabilność produktów pośrednich oraz ilość i krytyczny charakter odpadów procesowych, syntezy typu Koenigs-Knorr i inne techniki grup ochronnych stwarzałyby poważne problemy techniczne i ekonomiczne. Procesy typu Fischera są stosunkowo mniej skomplikowane i łatwiejsze do przeprowadzenia na skalę komercyjną, a zatem są preferowaną metodą produkcji alkilopoliglikozydów na dużą skalę.
Czas publikacji: 12 września 2020 r.